This web page was produced as an assignment for Genetics 564, an undergraduate course at UW-Madison.
What is Lesch-Nyhan Syndrome?
Lesch-Nyhan Syndrome (LNS) is a lethal, X-linked recessive disease caused by a severe deficiency of hypoxanthine-guanine phosphoribosyltransferase (HPRT), an enzyme that recycles purines, one group of building blocks for DNA [1]. The HPRT enzyme is encoded by the hypoxanthine phosphoribosyltransferase 1 (HPRT1) gene. The deficiency in purine recycling causes the overproduction of uric acid and a spectrum of clinical problems that includes neurological and motor development problems as well as self-mutilative behaviors [2].
From YouTube 2013. Patient with Lesch-Nyhan Syndrome. Retrieved from: https://www.youtube.com/watch?v=1U6LDpF_LFE
Clinical Signs and Symptoms
All people with LNS lack the wild-type HPRT enzyme. This leads to hyperuricemia and hyperuricosuria- the overproduction of uric acid in the blood and urine, respectively. Excessive uric acid crystallizes and is deposited in the joints between bones, causing irritation and a form of arthritis known as gout (Figure 1). Uric acid can also accumulate in the kidneys to form kidney stones that cause pain, block urine flow, and lead to renal failure (Figure 2) [3,4].

Figure 1: Uric acid crystals are deposited in joints, causing swelling, pain, irritation, and gouty arthritis.
|
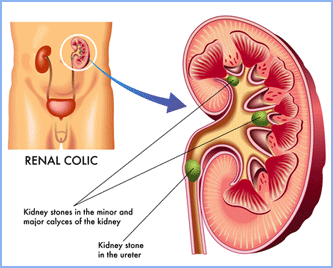
Figure 2: Uric acid hardens into kidney stones, which can block urine flow, are painful, and can cause renal failure.
|
People with Lesch-Nyhan Syndorme are born with the disease; it cannot be developed later in life. One of the first signs of LNS is a delay in psychomotor abilities within 3-6 months of birth as well as the presence of orange crystals in diapers. The following neurological symptoms of LNS are highly variable and are based on the type of mutation and degree of HPRT1 deficiency in each patient. The symptoms fall under the categories of motor control, cognition, and self-injurious behavior [5].
Motor Control
Patients with LNS experience involuntary muscle contractions. One type, dystonia, causes "involuntary muscle contractions that cause slow repetitive movements or abnormal postures" [6]. This can lead LNS patients to be unable to stand up or walk, confining them to a wheelchair. Other involuntary muscle contractions include arm flailing (ballismus) and jerking movements (chorea) [2].
Cognition
Patients with LNS exhibit varying levels of mental retardation, but most patients fall on the mild-to-moderate end of the spectrum. Most patients show attention deficit disorders but have normal non-verbal intelligence levels [7].
Self-Injurious Behavior
Compulsive self-mutilation is the most bizarre element of LNS, and it only occurs in patients that lack all HPRT enzyme activity. Patients usually begin by biting their lips, tongue, and fingers, creating self-wounds that cause significant pain (Figures 3,4). This obsessive-compulsive behavior starts between 2-16 years old and is often triggered by stress. Patients may also bite other parts of their bodies or bang their heads to injure themselves. Many patients direct their aggression toward caregivers through spitting and inappropriate language. When not attempting to injure themselves, LNS patients are often happy and engaging people [8].
Patients with all of these symptoms have classical Lesch-Nyhan Syndrome (LNS). Patients that have hyperuricemia and neurological issues, but no behavioral problems, are said to have 'HPRT-related hyperuricemia with neurological dysfunction' (HRND). Finally patients with hyperuricemia, but no neurological or behavioral issues, have 'HPRT-related hyperuricemia' (HRH) [1].
Motor Control
Patients with LNS experience involuntary muscle contractions. One type, dystonia, causes "involuntary muscle contractions that cause slow repetitive movements or abnormal postures" [6]. This can lead LNS patients to be unable to stand up or walk, confining them to a wheelchair. Other involuntary muscle contractions include arm flailing (ballismus) and jerking movements (chorea) [2].
Cognition
Patients with LNS exhibit varying levels of mental retardation, but most patients fall on the mild-to-moderate end of the spectrum. Most patients show attention deficit disorders but have normal non-verbal intelligence levels [7].
Self-Injurious Behavior
Compulsive self-mutilation is the most bizarre element of LNS, and it only occurs in patients that lack all HPRT enzyme activity. Patients usually begin by biting their lips, tongue, and fingers, creating self-wounds that cause significant pain (Figures 3,4). This obsessive-compulsive behavior starts between 2-16 years old and is often triggered by stress. Patients may also bite other parts of their bodies or bang their heads to injure themselves. Many patients direct their aggression toward caregivers through spitting and inappropriate language. When not attempting to injure themselves, LNS patients are often happy and engaging people [8].
Patients with all of these symptoms have classical Lesch-Nyhan Syndrome (LNS). Patients that have hyperuricemia and neurological issues, but no behavioral problems, are said to have 'HPRT-related hyperuricemia with neurological dysfunction' (HRND). Finally patients with hyperuricemia, but no neurological or behavioral issues, have 'HPRT-related hyperuricemia' (HRH) [1].

Figure 3: LNS patients often bite off fingernails.
|

Figure 4: Some LNS patients bite off tips of fingers.
|
The mechanism in which an HPRT deficiency leads to the neurological problems associated with LNS is currently unclear. Some human studies have shown dopaminergic system malfunction in the brain of Lesch-Nyhan patients [9]. One pharmacological rat model of Lesch-Nyhan showed that rats displayed self-injurious behavior in response to deficient dopamine levels [10]. It is unclear how the purine metabolic disorder leads to a dopamine deficiency in the brain.
Inheritance of Lesch-Nyhan Syndrome
|
LNS is an X-linked recessive disease (Figure 5). Mutations in the HPRT1 gene on the X-chromosome render the HPRT enzyme defective, which leads to disease. Females (XX) contain two X-chromosomes. A woman that carries a mutated HPRT1 gene on one X-chromosome will not manifest LNS because her other X-chromosome encodes the wild-type HPRT enzyme. Therefore, most women are carriers and do not show clinical signs of the disease unless they have mutated HPRT1 genes on both X-chromosomes, which is very rare [1].
Men (XY), have only one X-chromosome. Therefore, if a man inherits a defective copy of the HPRT1 gene from his mother, then he will have LNS. There is no normal copy of the gene to "cover up" the mutated copy. For this reason, LNS is found almost exclusively in men. The prevalence of the disease is 1/380,000 live births in Canada and 1/235,000 live births in Spain [1]. For comparison, a man born in Canada and living 80 years in the U.S. is ~32 times more likely to be hit by lightning over his lifetime than to be born with LNS [11]. |

Figure 5: An example of an X-linked disease, like LNS, in which the carrier mother gives the disease to her son.
|
The HPRT Enzyme and the Purine Salvage Pathway
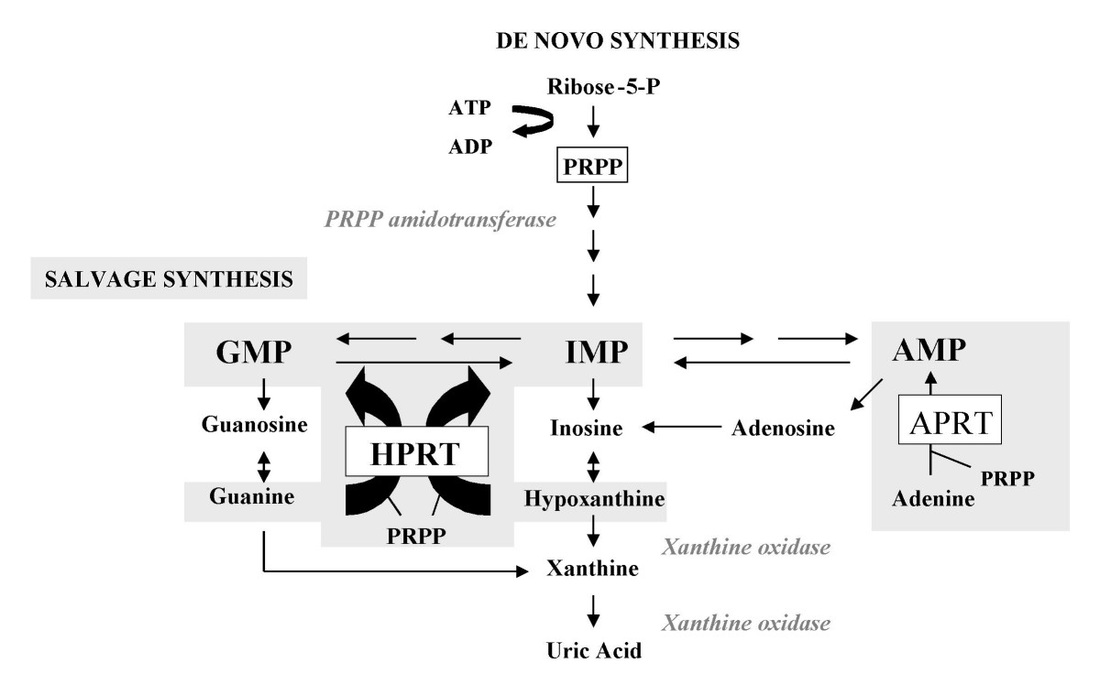
Figure 6: The de novo synthesis and salvage pathways for purine biosynthesis. HPRT is an enzyme that utilizes the phosphoribosyl-pyrophosphate (PRPP) co-substrate to recycle hypoxanthine and guanine bases back into inosine monophosphate (IMP) and guanine monophosphate (GMP), respectively. When HPRT is deficient, guanine and inosine are broken down into xanthine and then uric acid, which accumulates in the blood and urine.
Guanine, adenine, xanthine, and hypoxanthine are purines. Purines are important building blocks for DNA and RNA. While de novo purine biosynthesis is energetically costly, the purine salvage pathway efficiently recycles guanine and hypoxanthine back into GMP and IMP, respectively, using the HPRT enzyme (Figure 6). In LNS patients, the HPRT1 gene is mutated such that the HPRT enzyme is lacking in function or otherwise deficient. When this happens, guanine and hypoxanthine accumulate and are not recycled back into GMP and IMP. Instead, all guanine and hypoxanthine is converted into xanthine and then uric acid by the xanthine oxidase enzyme. Since guanine and hypoxanthine are not recycled, more purines are created via de novo synethesis. The increased flux of purines through the pathway generates the elevated uric acid levels consistent with LNS [1].
HPRT1 Gene
The human HPRT1 gene is a single gene found on the long arm of the X chromosome, at position Xq26 (Figure 7). The entire gene spans 45 Kb. However, the coding sequence contains just nine exons, with a total length of 1,435 bp [accession number: NM_000194.2, FASTA]. The HPRT1 gene codes for a 218 amino acid protein [accession number: NP_000185.1, FASTA].

Figure 7: Human chromosome X. The yellow arrow indicates the location of the HPRT1 gene (location Xq26).
Most people with LNS still present HPRT1 mRNA expression, so molecular diagnosis can occur via cDNA sequencing [12]. The types of mutations in the HPRT1 gene in LNS patients varies widely in both type and location within the gene. HPRT deficiency has been shown to be caused by point mutations, duplication, insertions, and deletions at various points within the HPRT1 gene. More than 300 different mutations have been found to cause LNS to date [13]. Single point mutations generally cause partial HPRT deficiency, but mutations that modify the size, shape, and therefore function of the wild-type HPRT enzyme cause LNS [14].
Diagnosis and Treatment
Patients with hyperuricemia and uric acid overproduction should be tested for an HPRT deficiency in conjunction with delays in psychomotor development. Generally, an HPRT activity assay is used to determine if the enzyme is functional. In women carriers, however, the wild-type HPRT masks the deficient HPRT enzyme. Both women carriers and men with LNS have elevated levels of uric acid, which can be tested using a urine sample. A diagnosis of LNS can be made as early as 10-12 weeks into gestation by collecting chorionic villus cells and testing for HPRT enzymatic activity [1].
Allopurinol is a xanthine oxidase inhibitor, meaning it prevents the production of uric acid from xanthine and hypoxanthine precursors [15]. Allopurinol reduces the amount of uric acid in the blood and urine, thereby preventing crystal formation, gouty arthritis, and kidney stone formation. The rate of hypoxanthine and xanthine excretion is 5-10 times greater in patients treated with allopurinol compared with controls. The reduction of uric acid levels using allopurinol does not ameliorate the behavioral and neurological symptoms associated with LNS. Drugs such as benzodiazepines can be used to relieve anxiety and manage dystonia [16]. Restraints are often needed to prevent LNS patients from harming themselves, and patients' teeth are often removed to prevent them from biting off their lips, tongue and fingers. With living assistance, patients with LNS generally live into their 20's or 30's.
Allopurinol is a xanthine oxidase inhibitor, meaning it prevents the production of uric acid from xanthine and hypoxanthine precursors [15]. Allopurinol reduces the amount of uric acid in the blood and urine, thereby preventing crystal formation, gouty arthritis, and kidney stone formation. The rate of hypoxanthine and xanthine excretion is 5-10 times greater in patients treated with allopurinol compared with controls. The reduction of uric acid levels using allopurinol does not ameliorate the behavioral and neurological symptoms associated with LNS. Drugs such as benzodiazepines can be used to relieve anxiety and manage dystonia [16]. Restraints are often needed to prevent LNS patients from harming themselves, and patients' teeth are often removed to prevent them from biting off their lips, tongue and fingers. With living assistance, patients with LNS generally live into their 20's or 30's.
Sources Cited
[1] Torres RJ and Puig JG. (2007). Hypoxanthine-guanine phosophoribosyltransferase (HPRT) deficiency: Lesch-Nyhan syndrome. Orphanet Journal of Rare Diseases, 2, 48. doi:10.1186/1750-1172-2-48
[2] "Lesch-Nyhan syndrome". Genetics Home Reference. US National Library of Medicine, Feb. 2013. Web. 24 Jan. 2015. <http://ghr.nlm.nih.gov/condition/lesch-nyhan-syndrome>
[3] "Lesch-Nyhan Syndrome". NYU Langone Medical Center. Aug. 2014. Web. 25 Jan. 2015. <http://www.med.nyu.edu/content?ChunkIID=22829#prevention>
[4] "Gout". Mayo Clinic. 25 Nov. 2014. Web. 24 Jan. 2015. <http://www.mayoclinic.org/diseases-conditions/gout/basics/definition/con-20019400>
[5] García PJ, Torres JR, Mateos F, Ramos T, Arcas J, Buño A, O'Neill P. (2001). The spectrum of Hypoxanthine-Guanine Phosphoribosyltransferase (HPRT) deficiency. Clinical experience based on 22 patients from 18 Spanish families. Medicine (Balt), 80, 102-112.
[6] "Dystonias Fact Sheet". National Institute of Neurological Disorders and Stroke. 12 Nov. 2014 Web. 24 Jan. 2015. <http://www.ninds.nih.gov/disorders/dystonias/detail_dystonias.htm>
[7] Schretlen DJ, Harris JC, Park KS, Jinnah HA, del Pozo NO. (2001). Neurocognitive functioning in Lesch-Nyhan disease and partial hypoxanthine-guanine phosphoribosyltransferase deficiency. J Int Neuropsychol Soc, 7, 805-812.
[8] Anderson LT, Ernst M. (1994). Self-injury in Lesch-Nyhan disease. J Autism Dev Disord, 24, 67-81.
[9] Ernst M, Zametkin AJ, Matochik JA, Pascualvaca D, Jons PH, Hardy C, Hankerson JG, Doudet DJ, Cohen RM. (1996). Presynaptic dopaminergic deficits in Lesch-Nyhan disease. New Engl. J Med, 334, 1568-1572. DOI: 10.1056/NEJM199606133342403
[10] Breese GR, Criswell HE, Duncan GE, Mueller RA. (1990). A dopamine deficiency model of Lesch-Nyhan disease – the neonatal-6-OHDA-lesioned rat. Brain Res Bull, 25, 477-484. DOI: 10.1016/0361-9230(90)90240-Z
[11] "How Dangerous Is Lightning?". Lightning Safety. National Weather Service. Web. 24 Jan. 2015. <http://www.lightningsafety.noaa.gov/odds.htm>
[12] Davidson BL, Tarle SA, Palella TD, Kelley WN. (1989). Molecular basis of hypoxanthine-guanine phosphoribosyltransferase deficiency in ten subjects determined by direct sequencing of amplified transcripts. J Clin Invest, 84, 342-346.
[13] Jinnah HA, De Gregorio L, Harris JC, Nyhan WL, O'Neill JP. (2000). The spectrum of inherited mutations causing HPRT deficiency: 75 new cases and a review of 196 previously reported cases. Mutat Res, 463, 309-326. doi: 10.1016/S1383-5742(00)00052-1
[14] Torres RJ, Mateos FA, Molano J, Gathoff BS, O'Neill JP, Gundel RM, Trombley L, Puig JG. (2000). Molecular basis of hypoxanthine-guanine phosphoribosyltransferase deficiency in thirteen Spanish families. Hum Mutat,15, 383.
[15] Rundles RW. (1985). The development of allopurinol. Arch Intern Med, 145, 1492-1503.
[16] Olson L, Houlihan D. (2000). A review of behavioral treatments used for Lesch-Nyhan syndrome. Behav Modif, 24, 202-222.
Media Sources
YouTube Video: https://www.youtube.com/watch?v=1U6LDpF_LFE
Inheritance picture: http://disorders.eyes.arizona.edu/sites/disorders.eyes.arizona.edu/files/XLinked_R_M.png
Gout picture: http://www.gnet.org/wp-content/uploads/explained-got-disease.jpg
Kidney stones picture: http://www.keystonekidney.com/images/about-kidney-stones.gif
Fingernails: http://simple-health-secrets.com/wp-content/uploads/2013/06/Lesch-Nyhan-Syndrome-4.jpg
Fingers: http://medicalpictures.net/wp-content/uploads/2010/10/Lesch-Nyhan-Syndrome-pictures-3.jpg
Purine Salvage Pathway: http://www.ojrd.com/content/2/1/48/figure/F1
Gene position: http://wiki.ggc.usg.edu/images/8/8a/HPRT1.jpg
[1] Torres RJ and Puig JG. (2007). Hypoxanthine-guanine phosophoribosyltransferase (HPRT) deficiency: Lesch-Nyhan syndrome. Orphanet Journal of Rare Diseases, 2, 48. doi:10.1186/1750-1172-2-48
[2] "Lesch-Nyhan syndrome". Genetics Home Reference. US National Library of Medicine, Feb. 2013. Web. 24 Jan. 2015. <http://ghr.nlm.nih.gov/condition/lesch-nyhan-syndrome>
[3] "Lesch-Nyhan Syndrome". NYU Langone Medical Center. Aug. 2014. Web. 25 Jan. 2015. <http://www.med.nyu.edu/content?ChunkIID=22829#prevention>
[4] "Gout". Mayo Clinic. 25 Nov. 2014. Web. 24 Jan. 2015. <http://www.mayoclinic.org/diseases-conditions/gout/basics/definition/con-20019400>
[5] García PJ, Torres JR, Mateos F, Ramos T, Arcas J, Buño A, O'Neill P. (2001). The spectrum of Hypoxanthine-Guanine Phosphoribosyltransferase (HPRT) deficiency. Clinical experience based on 22 patients from 18 Spanish families. Medicine (Balt), 80, 102-112.
[6] "Dystonias Fact Sheet". National Institute of Neurological Disorders and Stroke. 12 Nov. 2014 Web. 24 Jan. 2015. <http://www.ninds.nih.gov/disorders/dystonias/detail_dystonias.htm>
[7] Schretlen DJ, Harris JC, Park KS, Jinnah HA, del Pozo NO. (2001). Neurocognitive functioning in Lesch-Nyhan disease and partial hypoxanthine-guanine phosphoribosyltransferase deficiency. J Int Neuropsychol Soc, 7, 805-812.
[8] Anderson LT, Ernst M. (1994). Self-injury in Lesch-Nyhan disease. J Autism Dev Disord, 24, 67-81.
[9] Ernst M, Zametkin AJ, Matochik JA, Pascualvaca D, Jons PH, Hardy C, Hankerson JG, Doudet DJ, Cohen RM. (1996). Presynaptic dopaminergic deficits in Lesch-Nyhan disease. New Engl. J Med, 334, 1568-1572. DOI: 10.1056/NEJM199606133342403
[10] Breese GR, Criswell HE, Duncan GE, Mueller RA. (1990). A dopamine deficiency model of Lesch-Nyhan disease – the neonatal-6-OHDA-lesioned rat. Brain Res Bull, 25, 477-484. DOI: 10.1016/0361-9230(90)90240-Z
[11] "How Dangerous Is Lightning?". Lightning Safety. National Weather Service. Web. 24 Jan. 2015. <http://www.lightningsafety.noaa.gov/odds.htm>
[12] Davidson BL, Tarle SA, Palella TD, Kelley WN. (1989). Molecular basis of hypoxanthine-guanine phosphoribosyltransferase deficiency in ten subjects determined by direct sequencing of amplified transcripts. J Clin Invest, 84, 342-346.
[13] Jinnah HA, De Gregorio L, Harris JC, Nyhan WL, O'Neill JP. (2000). The spectrum of inherited mutations causing HPRT deficiency: 75 new cases and a review of 196 previously reported cases. Mutat Res, 463, 309-326. doi: 10.1016/S1383-5742(00)00052-1
[14] Torres RJ, Mateos FA, Molano J, Gathoff BS, O'Neill JP, Gundel RM, Trombley L, Puig JG. (2000). Molecular basis of hypoxanthine-guanine phosphoribosyltransferase deficiency in thirteen Spanish families. Hum Mutat,15, 383.
[15] Rundles RW. (1985). The development of allopurinol. Arch Intern Med, 145, 1492-1503.
[16] Olson L, Houlihan D. (2000). A review of behavioral treatments used for Lesch-Nyhan syndrome. Behav Modif, 24, 202-222.
Media Sources
YouTube Video: https://www.youtube.com/watch?v=1U6LDpF_LFE
Inheritance picture: http://disorders.eyes.arizona.edu/sites/disorders.eyes.arizona.edu/files/XLinked_R_M.png
Gout picture: http://www.gnet.org/wp-content/uploads/explained-got-disease.jpg
Kidney stones picture: http://www.keystonekidney.com/images/about-kidney-stones.gif
Fingernails: http://simple-health-secrets.com/wp-content/uploads/2013/06/Lesch-Nyhan-Syndrome-4.jpg
Fingers: http://medicalpictures.net/wp-content/uploads/2010/10/Lesch-Nyhan-Syndrome-pictures-3.jpg
Purine Salvage Pathway: http://www.ojrd.com/content/2/1/48/figure/F1
Gene position: http://wiki.ggc.usg.edu/images/8/8a/HPRT1.jpg
Website Creator: Billy Maes
Last Updated: May 7, 2015
www.genetics564.weebly.com
University of Wisconsin - Madison
Last Updated: May 7, 2015
www.genetics564.weebly.com
University of Wisconsin - Madison

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}